
Pesquisa conduzida por cooperação Brasil-Alemanha busca compreender reações eletroquímicas
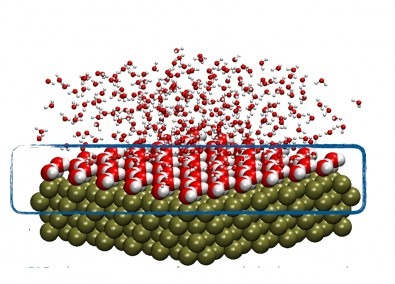
Simulação atomística da interface entre um metal (esferas verdes) e a água (corpúsculos vermelhos e brancos. (imagem: Luana Sucupira Pedroza)
A necessidade de produzir energia mais limpa, no contexto da crise climática global, é a motivação de uma pesquisa inovadora apresentada no evento Frontiers of Science Symposium, organizado pela FAPESP e pelo Instituto Max Planck.
O estudo, conduzido por Luana Sucupira Pedroza, professora da Universidade Federal do ABC (UFABC), visa otimizar reações eletroquímicas em células a combustível e outros dispositivos. Essas reações, cujo produto principal de interesse é o hidrogênio, envolvem a interação sólido-líquido. E a pesquisa busca justamente entender, por meio de simulação computacional, o que ocorre na interface dos dois sistemas.
O estudo conta com auxílio da FAPESP por meio do programa Apoio a Jovens Pesquisadores e de convênio com a Sociedade Max Planck para o Avanço da Ciência, da Alemanha.
Simulação atomística da interface entre um metal (esferas verdes) e a água (corpúsculos vermelhos e brancos. (imagem: Luana Sucupira Pedroza)
“Embora a eletroquímica seja conhecida há muito tempo, ainda não compreendemos completamente como a interface sólido-líquido se comporta na escala microscópica e o que podemos fazer nela para melhorar a eficiência da reação. O objetivo de nossa pesquisa é, basicamente, entender o que acontece na interface entre um sólido e um líquido – em particular, a água”, disse Pedroza à Agência FAPESP.
Além de contribuir para o desenvolvimento da ciência fundamental, explicando o que ocorre no contexto experimental, o estudo tem um evidente horizonte de aplicação tecnológica, possibilitando escolher os melhores materiais sólidos ou projetar novas configurações de interface.
“Trabalhamos no âmbito teórico, com simulação computacional, mas estamos muito conectados com a pesquisa experimental. Temos uma colaboração com físicos experimentais da Alemanha e as nossas simulações visam, entre outras coisas, explicar os resultados que eles obtêm, mas nem sempre conseguem compreender. As simulações atomísticas são indispensáveis para esse tipo de explicação”, afirmou a pesquisadora.
“O nosso estudo envolve as chamadas simulações ab initio – isto é, de primeiros princípios. Isso significa que não utilizamos os dados experimentais para montar nossos modelos. Usamos o conhecimento dos processos físicos fundamentais e os recursos de cálculo. É claro que sempre são necessárias aproximações, mas o objetivo é descrever os fenômenos reais com o mínimo de pressupostos, de modo a não confinar a interpretação no repertório empírico já estabelecido. Queremos ser capazes de prever novas possibilidades”, acrescentou.
Isso tem a ver, é claro, com o material sólido utilizado. A pesquisadora tem estudado principalmente paládio, platina e ouro – metais de interesse tanto como eletrodos quanto como catalisadores. Mas, além do tipo de material, o comportamento da interface também depende da geometria. “A interação com a água muda de acordo com a forma como o material é cortado, isto é, de acordo com os planos de clivagem. O comportamento da água não depende apenas do metal. Depende também da geometria da superfície metálica. Isso é algo muito importante que as simulações possibilitam explicar. A configuração das moléculas de água na interface é condicionada pela geometria da superfície”, disse Pedroza.
A molécula de água (H2O) é coplanar – os centros do átomo de oxigênio e dos dois átomos de hidrogênio localizam-se no mesmo plano. E, nesse plano, os centros posicionam-se na forma de um V, com o oxigênio no vértice e os hidrogênios nas duas pontas, formando um ângulo de aproximadamente 104,5 graus entre si. Dependendo do metal e da geometria da superfície metálica, isto é, das coordenadas do plano de clivagem, as moléculas da primeira camada de água podem ficar paralelas à superfície ou girar, de modo que os hidrogênios se aproximem ou se afastem da superfície. Essas diferentes configurações influenciam fortemente a reação.
O fato de os átomos da molécula da água formarem um V faz com que a molécula da água se constitua como um dipolo elétrico. Várias propriedades da água – inclusive o fato de ela ser um ótimo solvente – decorrem desse dipolo. Não é o que acontece, por exemplo, com a molécula do dióxido de carbono (CO2). Neste caso, os três átomos se alinham, o carbono do centro e um oxigênio em cada ponta, o que impede a polarização.

“As propriedades da água dependem também do ordenamento local do líquido. Isto é, de como cada molécula se relaciona com as moléculas vizinhas. Esse comportamento depende, por um lado, das relações carga-carga, o que constitui um componente clássico, puramente eletrostático. Mas depende também, por outro lado, das relações entre os orbitais eletrônicos dos átomos, o que determina um componente quântico. Nas simulações ab initio, os núcleos atômicos são tratados como corpúsculos clássicos e orbitais eletrônicos como sistemas quânticos. Este é o procedimento-padrão adotado desde o final dos anos 1980. Mas, como o hidrogênio é um átomo muito leve, tal procedimento nem sempre constitui uma boa aproximação com o fenômeno real. E aqui entra uma novidade do nosso estudo, que é incluir efeitos quânticos também para os núcleos. Este é o principal aspecto da nossa colaboração com a Alemanha. Nossa parceira alemã é especialista nisso”, afirmou Pedroza.
Tornar tudo quântico em um modelo constituído por algumas centenas de moléculas, no qual coexistem e competem interações metal-água e água-água, implica utilizar as chamadas “integrais de caminho de Feynman” – uma técnica que demanda processamento computacional pesado, com muito tempo de uso de máquina. Por meio do programa Apoio a Jovens Pesquisadores, a FAPESP aportou a Pedroza recursos financeiros para isso. Além dos clusters de computadores da Universidade Federal do ABC (UFABC) e do Laboratório Nacional de Computação Científica (LNCC), em Petrópolis-RJ, que ela já utiliza em regimes de hora-máquina, com limites de tempo, a pesquisadora está em processo de compra de um cluster destinado a atender prioritariamente ao seu projeto de pesquisa. Esse novo cluster será sediado na UFABC.
Além de efeitos quânticos para os elétrons e para os núcleos, o modelo deverá incluir também efeitos de temperatura e efeitos de tensão elétrica externa aplicada ao metal. Isso demanda simulações realmente complexas, que disponibilizarão para a comunidade científica descrições bastante realísticas dos processos eletroquímicos. “Juntar todas essas variáveis é algo que ainda não foi feito. Nós esperamos fazer pela primeira vez”, comentou Pedroza.
Fonte: FAPESP


